

Бұл аурулар тобы гликолипидтер ыдырауы (цереброзид, ганглиозидтер) бұзылыстарымен ерекшеленеді. Гликолипидтер – церамид пен галактоза қосылыстарынан тұрады және құрамында (ганглиозидтер) нейрамин қышқылы мен гексозаминдер бар ете күрделі липоидтар. Осы қосылыстар метаболизмінің бұзылыстары түрлі тұқым қуалайтын арурулар туғызады. Гликолипидоздар негізінен аутосомды-рецессивті жолмен беріледі, бұл ереже тек Фабри синдромында өзгереді — мұнда ферменттік кемістік Ххромосомасымен қиылысады. Гликолипидоздар, цереброзидоздар (Гоше ауруы, Краббе ауруы), сульфатидоздар (метахроматикалық лейкодистрофия), церамидолигодоздар (церамидлактозидлипоидоз, Фабри синдромы), ганглиозидоздар: Тей-Сакс ауруы, Зандгофф-Яцкевич-Пильц ауруы, Бернгеймер-Зайтельберг ауруы, интфантильдік ганглиоздың ІV типі, Gm1– ганглиозидоздары: Норман – Ландиг ауруы, Дерри ауруы) болып ажыратылады.
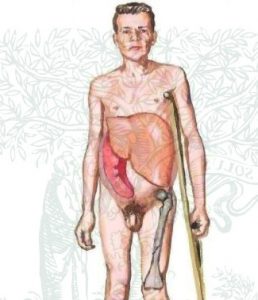
Гоше ауруы (глюкозилцерамидлипидоз ). Ауру негізінде глюкоцереброзидаза ферменті белсенділігінің жоғалуы жатады, бұл ретикулярлы жүйе (РЭЖ) жасушааларында глюкоцереброзид жиналуына әкеледі. Аурудың негізгі белгілеріне мұрын қанауы, терінің геморрагиялық бөртпелері, жатырдан көп қан кетуге бейімділік жатады. Өсуі, дамуы тоқтап, инфантилизм өрбіп, етеккірі кешігеді. Гоше ауруының айқын сипатына бауыр мен көк бауырдың ұлғаюы жатады, олардын көлемі орасан үлкен, іш аумағын түгелге жуық алады. Аурудын бұдан да басқа мәнді диагностикалық белгісі – сүйек жүйесінің өзгерісі. Қуысты сүйек ауырулары пайда болып, ол жүргенде күшейеді. Бас сүйектер бұзылысы болмайды.
Аурудың маңызды белгісіне (науқастардың жартысында байқалады) саусақтары мен беттің өзгеше қоңыр-қошқыл түс алып, оның дақты немесе диффузды түрде болуы жатады. Ауруларда түгелге дерлік гиперспленизм белгісі – анемия, лейкопения, тромбоцитопениялар – кездеседі. Аурудың негізгі үш типін ажыратады: созылмалы (І тип), бұған нерв жүйесі бұзылыстары тән емес, сондықтан кейде мұны үлкендер ауруы деп атайды; балалық немесе инфантильдік тип (ІІ тип) айқын нерв жүйесі бұзылыстарымен ерекшеленеді, бұны клиницистер жедел ағымды нейропатиялық ауруға жатқызады (бұлшыкеттер тонусының жоғарылауы, бұл соңынан опистотонус, клоникалық-тоникалық тырысу, тризмге апарады); бозбалалық немесе созылыңқы типі (Ш тип), ағым ауырлығына қарай бұл алғашқы екеуінің аралығында болады. Инфантильдік форманың өрбуі 6 айлық шамасында болады. Созылмалы типі ересек балаларда кездеседі. Аурудың өзіндік белгісіне тән мөлшерден тыс жиналған глюкоцереброзидтер гистиоциттерде жатады, олар анық (дөңгелек) формалы, Гоше жасушалары деп аталады. Көк бауырдың қызыл пульпасы, лимфа түйіндері, бауыр синусоиды мен сүйек миы бұл клеткалардың көптеп кездесетін орындары.
Сүйек миы тексерісінде Гоше клеткаларын табу – диагностика сынақтарының бірі. Соңғы кездері глюкоцереброзидазаны микроәдіспен анықтау ұсынылған. Фермент белсенділігі, лейкоциттерден өзге, бауыр, көк бауыр және тері фибробласттарында анықталуы мүмкін. Қан сүйығында қалыпты көрсеткішке қарағанда ауруларда қышқыл фосфатаза белсенділігі 5-50 есе артық.
Емі. Егер жалпы жағдайы ауырламай, анық спленомегалия, анемия мен геморрагия көріністері болмаса, емнің қажеті жоқ. Анық спленомегалия, сүйек бұзылыстары өрбіп, геморрагиялық синдром үдегенде, ең тиімді ем ретінде спленэктомия жасау мәселесі алға қойылады. Таңдаулы емге гормондар емі (преднизолон 1 мг/кг есебінен тәулігіне, кейін бірте-бірте азайтады), көк бауыр тұсына рентген сәулесін жасау, лейко-, тромбоцитопоэз жұмысын үстемелейтін дәрілер жатады (цередаза). Болжамы. Салыстырмалы түрде сәтті болып есептеледі.
Инфантилдік Gm2 ганглиозидоз (Тей-Сакс ауруы). Ауру негізінде генетикалык орайласқан ганглиозидтер алмасуы бұзылысы жатады. Осы себептен олар көп мөлшерде мидын бозғылт затына жиналады, бұл бауыр мен көк бауырға да қатысты. Тей-Сакс ауруында А-гексозаминидаза ферментінің кемістігі анықталады.
Инфантильдік ганглиозидоз жиілігі 1:250000, аутосомды-рецессивті жолмен беріледі. Туған және алғашқы 3-4 ай мерзімінде сәбилердің өз құрдастарынан өзгешелігі болмайды. Кесел баяу өрбіп, бала енжар тартады, үйренген дағдыларын ұмытып, ойынға зауқы болмай, анасын да танымайтын болады. Көру нашарлығы ерте байқалады (зер салып қарау, көрінген затқа ілесе қарау). Тез арада соқырлық дамиды, санырақулықтың да бірге өрбуі сирек емес. Сана бұзылыстары үдеп, аздан кейін ақыл-естің деградациясы идиотия дәрежесіне дейін барады. Гипотония пайда болып, бала басын да көтере алмайды, аяқ-қол салдануы байқалады. Тоникалық тырысу жиі көрініс алады. Псевдобульбарлық паралич көрінісі сирек емес: жұтыну бұзылысы, еріксіз күлу мен жылау. Балалар тез арықтап, 1-1,5 жаста өледі.
Диагнозы. Биохимиялық сынақ (гексозаминадаза белсенділігін анықтау), көз түбіндегі айқын езгерістер (көру нерві емізікшелерінің атрофиясы мен макула аймағындағы алқызыл шие тәрізді дақ) диагнозды қоюға арқау болады. Бұл кеселдің ұтымды емі жоқ. Тырысуға қарсы, жалпылай әл беретін дәрілер қолданылады.
Сфингомиелинлипидоз (Ниман-Пик ауруы) – жасушаішілік липоидоз, РЭЖ клеткаларында фосфолипид-сфингомиелин жиналуымен ерекшеленеді. Сфингомиелиндер алмасуының бұзылысы сфингомиелинидазаның ферменттік белсенділігін жогалтуына байланысты. Аутосомды-рецессивті жолмен беріледі, жиілігі 1:10000. Сфингомиелинлипидоз тек сәбилерде кездесіп, ағымы аса қатерлі болады. Ауру бастапқыда емшектен қалып, лоқсу, құсумен білінеді. Соңынан психо-моторлық қалысы байқалып, гепатоспленомегалия үдейді. Артынан спастикалық тетрапарез, соқырлық, саңыраулық пайда болады. Тері қоныр-қошқыл түстенеді. Көз түбінде жиі алқызыл шие тектес дақ көрінеді.
Ниман-Пик кеселін бірнеше клиникалык вариантка ажыратады: А, В, С және Д.
- А варианты – кеселдін классикалық формасы: нерв жүйесінің ауыр бұзылыстары айқын гепатоспленомегалиямен қатар орын алады;
- В вариантында нерв жүйесі кұбылысқа қатыспайды;
- С вариантына сәтті ағым тән, неврологиялық белгілер кешірек білінеді;
- Д вариантының ағымы тіптен өзгеше, созылыңқы гепатит бауыр циррозына ауысады.
Сфингомиелинлипидоз диагнозы сүйек миы мен көк бауыр пунктатынан Ниман-Пик клеткаларын табуға негізделеді. Олардың көлемі едәуір (90 мкм-ге дейін), спиртпен тұрақтандырғанда «көбік» түріне ұксас, себебі спирт цитоплазмадағы май тектес заттарды ерітеді. Бұл тектес клеткалар басқа да лизосомалық ауруларда табылады, мысалы, гиперлипидемия. Метаболикалық тосқауылдың энзимдік диагнозы ультрадыбыс әсерінен кейін тері фибробластар культурасынын сығындысы (экстракт) мен лейкоциттерді зертгеу арқылы анықталады.
Емі. Ниман-Пик кеселіне ем қонбайды. Липокаин, метионин, витаминдер қоспасын беруге болады. Сонғы кездерде липоидоздар емінің жана бағыттары қарастырылуда: мысалы, орын толтыру ретінде липосомалармен тазартылған ферменттік дәрілерді енгізу. Негізгі қиындық енгізілген ферменттік белок өмірінің қысқалығында және гематоэнцефальдық кедергінің алынбауы болып отыр. Вирустарды қолданып генетикалық трансдукция мүмкіндігі іздестірілуде: бұлар арқылы кеміс ген ауыстырылып немесе жоқ цистрон енгізіледі (цистрон полипептидте аминқышқылдарының арнайы тізбегінің орнын, бірлігін қадағалайтын жер – локус).